AstraZeneca says its COVID-19 vaccine needs ‘additional study’
UK drug company says that while further research is needed, it did not expect it to delay regulatory approval in Europe.

Published On 26 Nov 2020
AstraZeneca might have to run an additional global trial to assess the efficacy of its COVID-19 vaccine, after concerns were raised about the effectiveness of its jab.
The British company’s chief executive Pascal Soriot was quoted as saying in a Bloomberg News report on Thursday that an additional study would be run to evaluate a lower dosage that performed better than a full amount in AstraZeneca’s studies.
Keep reading
list of 3 itemsOxford-AstraZeneca COVID-19 vaccine results expected by Christmas
AstraZeneca chief says COVID vaccine on track for year end
“Now that we’ve found what looks like a better efficacy we have to validate this, so we need to do an additional study,” Soriot was quoted as saying.
Soriot said it would probably be another “international study, but this one could be faster because we know the efficacy is high, so we need a smaller number of patients”.
The news comes as AstraZeneca, which is working with the University of Oxford, has faced questions about its success rate. Some experts have said it could hinder its chances of getting speedy approval by regulators in the United States and European Union.
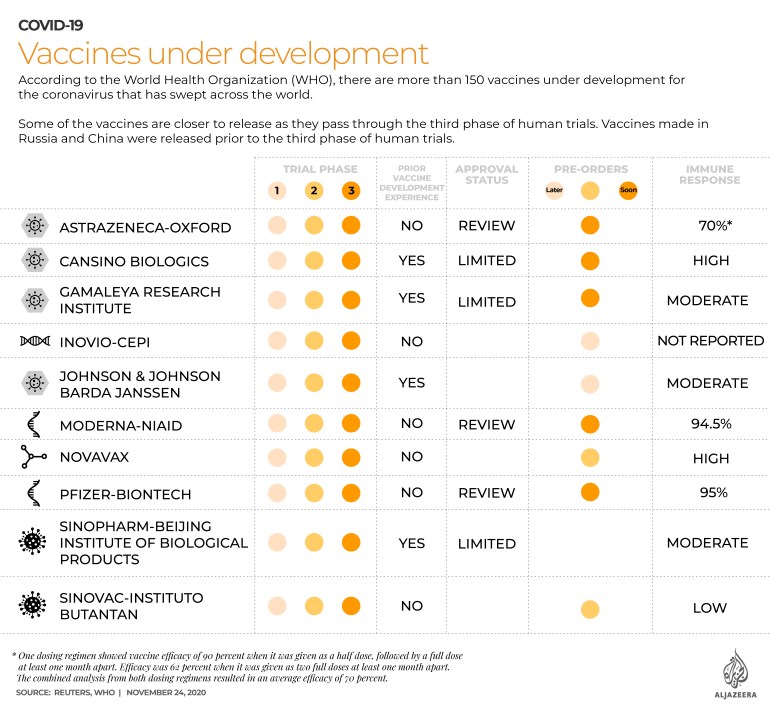
Several scientists have raised doubts about the robustness of results released on Monday showing the experimental vaccine was 90 percent effective in a sub-group of trial participants who, initially by error, received a half-dose followed by a full dose.
Soriot said he did not expect the additional trial to delay United Kingdom and European regulatory approvals.
On Friday, Britain’s Health Minister Matt Hancock said the UK government had requested the regulator to assess the vaccine, and hoped to begin the inoculation programme before Christmas.
“We have formally asked the regulator to assess the Oxford/AstraZeneca vaccine, to understand the data and determine whether it meets rigorous safety standards,” Hancock said in a statement. “This letter is an important step towards deploying a vaccine as quickly and as safely as possible.”
Earlier Soriot said while authorisation was likely to come from some countries before the end of the year, clearance from the US Food and Drug Administration (FDA) could take longer because the agency was unlikely to approve the vaccine based on studies carried out elsewhere, especially given the questions about the results.
AstraZeneca research chief Mene Pangalos told Reuters on Monday that researchers had stumbled upon the half-dose regime by accident, saying a sub-group of the trial was given a smaller initial dose by mistake.
Earlier he had said that the firm would start discussions with the FDA to change the design of its experimental COVID-19 vaccine trial to add the more-effective dosage regime.
‘The vaccine works’
While this could constitute a setback for the UK company, Chris Smith, consultant virologist with Cambridge University, said the error could actually turn out in favour of AstraZeneca.
“What they found … was that they had one group of individuals who had a 90 percent-plus response rate to their vaccine, and another group that responded a bit less well, down 60 or 70 percent,” Smith told Al Jazeera.
“Then, by analysing the data, they have found that individuals who got a smaller amount first and then a bigger dose next, actually responded better than people who got two higher doses,” he said.
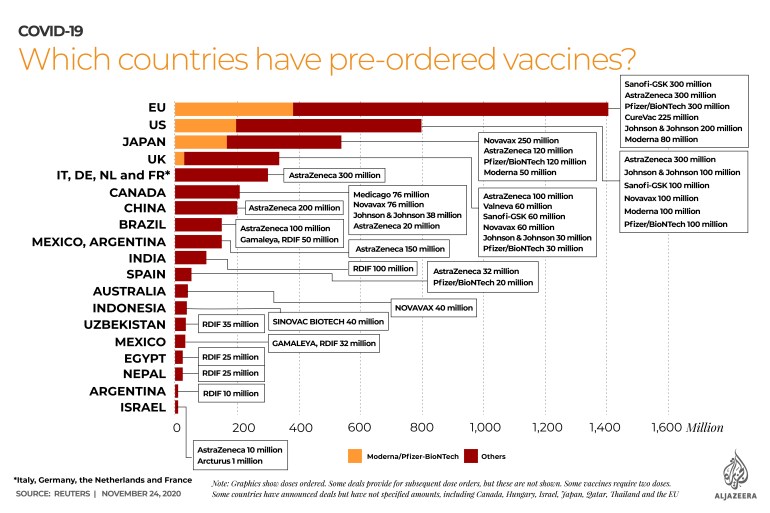
“If that turns out to be the case, then the 100 million doses of the vaccine that the UK has already purchased from AstraZeneca, instead of treating half of the population, will provide enough coverage to go through the entire population,” Smith added.
Meanwhile on the same day, the British government’s Chief Scientific Adviser Patrick Vallance said that the main point about the AstraZeneca vaccine against COVID-19 was that it worked, when asked about doubts that have been raised about the vaccine.
“The headline result is the vaccine works and that’s very exciting,” Vallance said during a news conference with Prime Minister Boris Johnson.
Chief Medical Adviser Chris Whitty, answering the same question, said there was always scientific debate about virtually everything.
“The key thing from our point of view is to leave this in the hands of the regulator … They will make an assessment with lots of data that is not currently in the public domain on efficacy and on safety,” Whitty said.
Source: Al Jazeera and news agencies